
Molecular and cellular effects of pathogenic NFKB1 and NFKB2 sequence variants in primary antibody deficiency syndromes
Common variable immunodeficiency (CVID) is the most prevalent symptomatic primary antibody deficiency (PAD), with recurrent infections, mostly in the upper respiratory tract. Genetic defects are identified in approximately 20 % of CVID patients. Among these, heterozygous pathogenic variants in NFKB1 or NFKB2 collectively account for up to 10 % of the monogenic forms. NFKB1 encodes the NF-kappaB1 (NF-κB1) transcription factor precursor p105 which is processed to p50 (canonical pathway), whereas NFKB2 encodes p100/p52 (non-canonical pathway). The dimeric transcription factors of the NF-κB family are composed of a variety of hetero- and homodimers of the subunits p50, p52 and the three Rel-proteins RelA (=p65), RelB and cRel, play important roles in numerous biological processes including the immune system and mediate transcriptional responses to a multitude of stimuli.
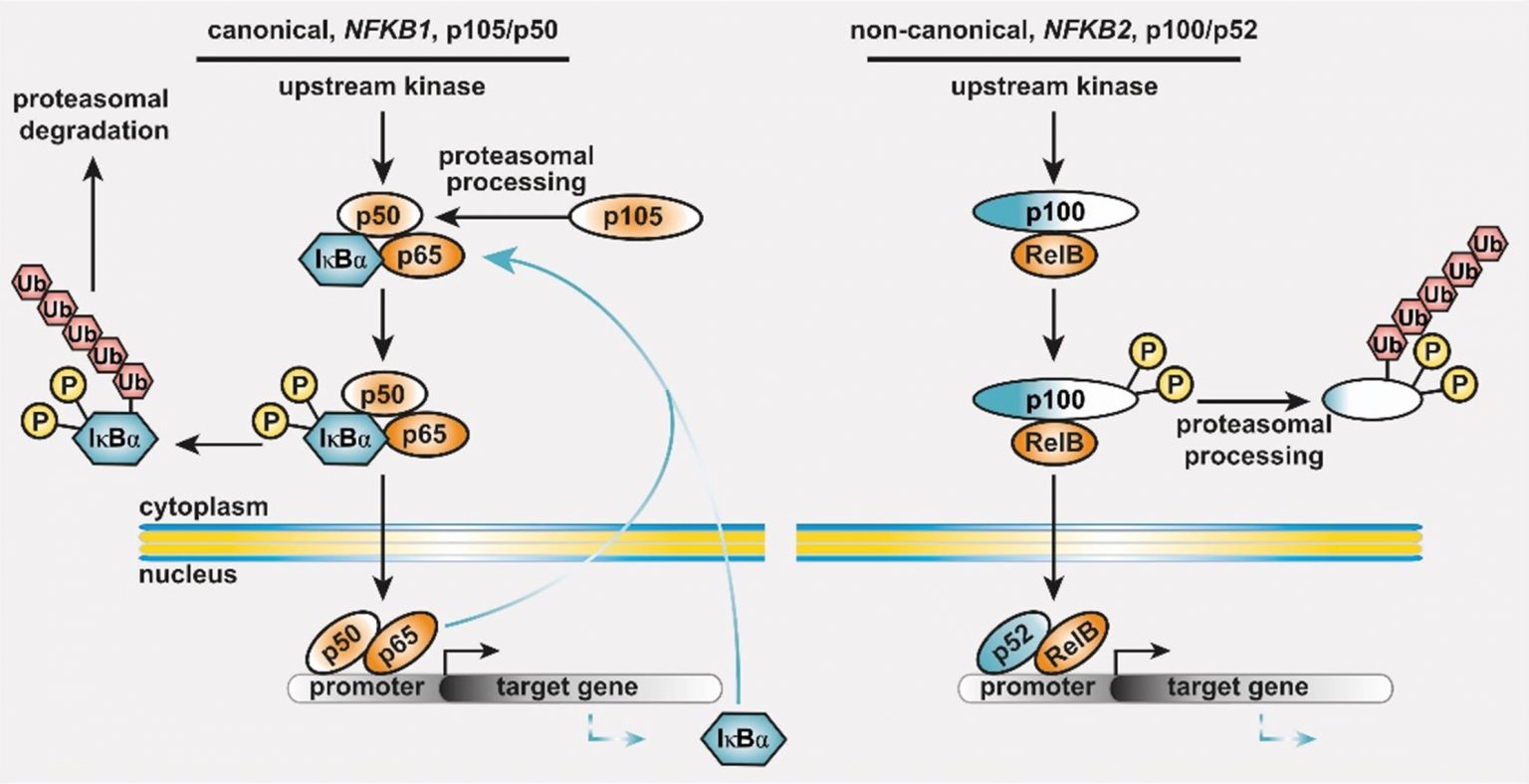
Figure 1: The basic principles of the NF-κB signaling systems.
The NF-κB signaling network is highly complex and operative in most cell types. (left) The canonical NF-κB1 pathway integrates signals from receptors (TCR, BCR, TNF-R and TLRs in lymphocytes), co-stimulators, and metabolites (e.g. glutamine). Processing of its C-terminal half converts the NF-κB1 precursor p105 into the transcription factor subunit p50, which assembles with RelA (also known as p65) and the inhibitory protein IκBα an inactive cytoplasmic trimer. Pathway stimulation activates upstream kinases, which leads to phosphorylation, polyubiquitination and degradation of IκBα. Subsequently, the preassembled transcription factor p50/RelA is released, translocates to the nucleus, and regulates its target genes. (right) Activation of the non-canonical NF-κB2 pathway occurs through BAFF receptor signaling, CD40 or the lymphotoxin-beta receptor. Pathway stimulation leads to proteasomal degradation of the inhibitory C-terminal half of the p100 precursor thereby releasing the active p52/RelB transcription factor. Homodimers of p50/p50 and p52/p52 are not equipped with a transactivation domain and act as transcriptional repressors (not shown). Both cytoplasmic precursor proteins administrate IκB-like activity on the transcription factor dimers.
In vitro analysis of protein defects associated with NFKB1 and NFKB2 sequence variants
We are using a cell-culture-based transient overexpression model to evaluate the pathogenic relevance of NFKB1 and NFKB2 sequence variants. Following site-directed mutagenesis, the ectopically expressed mutant proteins are analyzed for functional defects. The testing system solely relies on the knowledge of a given variant e.g. from genetic counseling, blood sampling is not required. According to the ‘type’ of the identified protein defect, we assign the variants into distinct categories.
The most-common ‘type’ of the known pathogenic NFKB1 variants affects the N-terminal half of the protein (typically on the N-terminal side relative to the nuclear localization sequence, NLS) and predicts the expression of severely truncated, non-functional proteins (if expressed at all) which undergo intensified decay and cause (haplo-)insufficiency of both, p105 and p50. Truncating variants in the central/proximal part of the protein predict skipping of the precursor stage and immediate expression of p50-like proteins. Rarely, truncating variants affect the C-terminal domains of p105, yet with unknown effects. Internal deletions result from splicing errors. A variety of known pathogenic missense variants (single amino acid changes), all residing within the N-terminal Rel-homology domain (RHD), cause protein-decay or, less frequent, loss-of-DNA-binding activity. The relevance of the frequently occurring missense variants within the C-terminal half of p105 (the part which is removed upon generation of p50) is largely unknown.
Most of the known disease-causing NFKB2 variants cause expression of non-processable p100 precursor proteins – and thus an excess of the associated IκB-like activity – and, as a ‘side-effect’, insufficiency of p52. Also known are (i) proximally truncating precursor-skipping variants predicting the expression of p52-like nuclear proteins, (ii) short truncations associated with haploinsufficiency of p100 and p52, and (iii) diverse missense variants with yet unpublished effects.

Figure 2: Pathogenic NFKB1 variants cause diverse protein defects.
Schematic protein structure of the p105 precursor with the N-terminal Rel homology domain (RHD), the central nuclear localization sequence (NLS) and the glycine-rich region (GRR), and the C-terminal Ankyrin-repeat domain (ANK) and the death domain (DD). The mature p50 comprises the N-terminal half of p105. Numbers indicate amino acid positions. Depending on the ‘type’ of protein defect, miscellaneous signaling processes might be deranged. Due to the complexity of NF-kB signaling, this concept is not exhaustive (adapted from Fliegauf & Grimbacher JACI 2018;142(4):1062-1065.)
NFKB1 and NFKB2-omics
The transcription factor NF-κB plays a pivotal role in the adaptive immune response. Pathogenic variants in NFKB1 are the most common genetic etiology of common variable immunodeficiency (CVID). Patients frequently present with impaired terminal B cell differentiation, autoimmunity, and hyperinflammatory immune dysregulation. NF-κB signaling and target gene expression are expected to be dysregulated in NFKB1-mutated patients. Here, we performed a multi-omics characterization of lymphocytes and monocytes from a cohort of clinically affected and unaffected NFKB1 mutation carriers. Our analysis of B lymphocytes identified specific epigenetic dysregulation and gene expression differences on B cells from NFKB1-mutated patients. We observed an aberrant expression of negative regulators of NF-κB signaling in NFKB1 mutation carriers, which may be a key factor for the autoinflammatory phenotype of these patients. Moreover, our analysis points towards a dysregulation of XBP1 and BCL3, key players of B cell activation and proliferation at different stages of B cell differentiation. The analysis of T cells and monocytes is still pending.
The reduced expression of negative regulators of the NF-κB network is likely to be one of several mechanisms responsible for the aberrant NF-κB signaling, which impairs the maintenance of a normal humoral immune response.
In summary, our findings highlight epigenetic and gene expression changes in B cells associated with NFKB1 mutations. Our data give insight into future therapeutic opportunities for patients with NFKB1 (haplo)insufficiency.
Phenotypic characterization, disease history, and treatment outcome of patients carrying pathogenic variants in NFKB1 or NFKB2
Analysis of the clinical and immunological phenotype of affected patients with pathogenic heterozygous NFKB1 variants has revealed an incomplete penetrance and an age-dependent disease severity. Patients are characterized by hypo-gammaglobulinemia, reduced switched memory B cells and recurrent respiratory and gastrointestinal infections. In addition, autoimmunity, lymphoproliferation, non-infectious enteropathy, opportunistic infections, autoinflammation and malignancy are frequently observed. Increased susceptibility to bacterial, viral and fungal infections is typical and autoantibodies are detectable in a rather large proportion.
Despite an unmet medical need, no targeted treatment is available for this rare condition. Current treatment protocols include immunoglobulin replacement and antibiotic treatment, as well as application of immunosuppressive or anti-inflammatory agents such as anti-IL1 and anti-TNF-α. Moreover, abatacept (a CTLA4 fusion protein) appears to be a promising option.
We therefore investigate, whether the observed autoinflammation and the increased frequency of viral infections (both reminiscent of interferonopathies) is associated with an altered type 1 interferon expression pattern in innate immune cells. Since the observed autoimmune disease is indicative of defective regulatory T cells (Tregs), we also aim at a detailed characterization and phenotyping of Tregs (and effector T cell subsets) in patients with pathogenic NFKB1 variants. In addition, we are testing whether the increased frequency of cytopenia in these patients is mediated by autoantibodies. We furthermore analyze whether a second genetic alteration (either somatic or germline) accounts for the observed incomplete and age-dependent penetrance.
To compare the effect of different therapeutic options, we have developed an NFKB1 Disease Activity Score (NFKB1-DAS). Using the score, the therapeutic success can be measured objectively in clinical trials. The score can also be used for clinical management to monitor the course of disease activity in patients. Based on data from the literature and data from patients in Germany and Switzerland, we decided which disease manifestations the score should reflect. The final score consists of 21 parameters, which depict the various organ manifestations of the disease, according to their severity. Further validation of the score is currently performed based on a global cohort of patients with NFKB1 insufficiency and its feasibility is being tested in practice. Treatment options such as anti-TNF-α, anti-IL1, or JAK-inhibition are currently being analysed using the NFKB1-DAS.
From many years of caring for our patients, we know that NFKB1 insufficiency may lead to a relevant impairment of quality of life. In our clinical research, we therefore implement questionnaires on quality of life and use these to evaluate the various treatment options, in addition to the NFKB1-DAS.
If you are interested in cooperating with us on our projects, for example by contributing patient data, or if you are interested in the NFKB1 Disease Activity Score, please contact Prof. Dr. Bodo Grimbacher.
The contributions of our group to the field of NF-κB research are the following:
Funding
This project is primarily funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy – CIBSS – EXC-2189 – Project ID 390939984.
Contact

Dr. Manfred Fliegauf
E-Mail
